
- Grayson Jones
Sickle Cell Disease (SCD) is inherited which means that the disease is passed by genes from parents to their children. People who have SCD inherit two abnormal hemoglobin genes, one from each parent. SCD has many forms; however, the most common and severe form, sickle cell anemia, overwhelming affects African-Americans and Hispanics in the United States. This paper will analyze SCD; explore the social implications and any genetic advantages; and report on the current societal implications.
Hemoglobin is a protein in red blood cells that carry oxygen throughout the body. Sickle Cell Disease (SCD) is a group of inherited red blood cell disorders which have abnormal hemoglobin. SCD is not contagious, like a cold or an infection, and is passed by one gene from each parent to their children. People with SCD have either one or two abnormal hemoglobin S genes. Sickle cell anemia is the disease that describes those with two hemoglobin S genes, hemoglobin SS.
Sickle cell trait is the condition where the hemoglobin S gene is inherited from one parent and a normal hemoglobin gene is inherited from the other parent. People with sickle cell trait are generally healthy and symptom free. Nevertheless, they are carriers of the defective hemoglobin S gene and can pass that defective gene to their children.
The National Heart, Lung, and Blood Institute’s (NHLBI) (2016) figure below shows the difference between normal and abnormal red blood cells. Normal red blood cells contain hemoglobin that is disc shaped which allow cells to provide a steady flow of oxygen to the body’s tissues. Abnormal red blood cells contain sickle hemoglobin which are not flexible and do not move freely to provide needed oxygen to the body’s tissues.
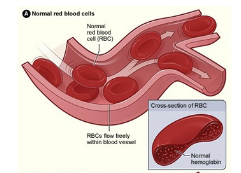
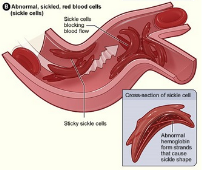
Lack of tissue oxygen causes attacks of severe and sudden pain. According to the NHLBI (2016), “Most children with SCD are pain free between painful crises, but adolescents and adults may also suffer with chronic ongoing pain. The red cell sickling and poor oxygen delivery can also cause organ damage. Over a lifetime, SCD can harm a person’s spleen, brain, eyes, lungs, liver, heart, kidneys, penis, joints, bones, or skin.”
Get Help With Your Essay
If you need assistance with writing your essay, our professional essay writing service is here to help!
Normal red blood cells live approximately 90 to 120 days while abnormal sickle cells typically last only 10 to 20 days. This is due to the fact that sickle cells cannot change shape easily and “burst apart or hemolyze” (NHLBI, 2016). The human body is continually reproducing new red blood cells to replace old blood cells which mean that a body with SCD has trouble keeping up with demand. The affect is a lower than normal red blood cell count called anemia.
The social implications of Sickle Cell Disease have been seen in a myriad of ways such as caregiving, community perceptions, and the health care system. Since SCD begins prior to birth and affects not only the children but the parents too, a rise in a family based approach to this disease has been seen. Mothers of a children with SCD are living in constant anxiety and stress that their child may become deathly ill at any moment, “intense, complex nature of SCD crises as unpredictable, recurring, and potentially severe” (Burnes, Antle, Williams, & Cook, 2008). The disease can arise and be triggered by a fluctuation in temperatures, stress, lack of sleep, and other factors. Since the symptoms come on suddenly and intensely, then this makes it difficult to predict onset which cause families to live in constant state of fear. In the study, the mothers felt that they are left with all the responsibility and to be the overall caregiver, while the fathers distance themselves from the child’s pain.
Another social implication of SCD is the community perception or lack of knowledge of the disease. SCD has not had a significant public awareness in the developing countries where most cases are prevalent, “Most mothers had not even heard of SCD, and they did not know how the illness is transmitted” (Burnes et al., 2008). The understanding is that women and men are not tested for the sickle cell trait and are not aware of how the disease is spread which has led to stigma about SCD in their cultures. There are many falsehoods about the disease such as contagious, a curse on one’s family, or being at fault for having a child who is born with such a physical illness. In addition, a racist assumption of the disease exists and a feeling of being powerless to speaking up about the disease because one does not want to be thought of less than anyone else.
The last social implication is what is identified in the health care system. There have been advancements in the treatment of SCD in first world countries, through medication and other treatments; however, there is still a lack of treatment possibilities in the areas that are mostly affected by SCD. The overall complaint among families affected by the disease is that there is a lack of knowledge among medical professionals, “did not know about SCD or how to treat its symptoms during a crisis” (Burnes et al., 2008). Parents going into a hospital and having to tell the staff about treatments is scary. They think that they cannot trust professionals to take care of their child since these professionals lack the required education about such a serious disease. The overall leading social implication of SCD is the lack of knowledge and education that is associated with this disease.
The Sickle Cell gene has a genetic protection against Malaria, which is a serious and sometimes deadly infectious disease. Malaria is caused by a mosquito that is carrying a parasite harmful to humans and the mosquito bites the human. Symptoms include high fevers, chills, and other flu like signs. If a person who is a carrier of one sickle cell gene (heterozygous), has shown signs of lower mortality and morbidity rate among those who may become infected with malaria, “Unexpectedly, heterozygous individuals experience some protection from malaria infections. Aidoo et al.(2002), demonstrate reduced mortality and morbidity, Aluoch 1997 reports higher resistance to malaria, whereas Hesran et al.(1999), demonstrate a reduced parasite load for heterozygous carriers of this otherwise damaging gene” (Lidell, Oswusu-Brackett, Wallace, 2014). Heterozygote Protection is when an individual who carries a normal allele and an infected allele are at an advantage and maintain that heterozygote presence in population. This is seen directly, “When the malarial protozoan invades the red blood cells of heterozygotes, the parasites cause a relatively large reduction in the oxygen tension within the cells and thus contribute to sickling. The sickling of the red blood cells then impairs the protozoan growth and development” (Howe, 2007). This means that the sickled blood cells stop the infection of malaria spreading throughout an individual; the heterozygotes are protecting the body from an infectious disease.
The exact number of people living with Sickle Cell Disease (SCD) in the United States (US) is not known. According to the Centers for Disease Control and Protection (CDC) (2016), “SCD affects approximately 100,000 Americans. SCD occurs among about 1 out of every 365 Black or African-American births. SCD occurs among about 1 out of every 16,300 Hispanic-American births. About 1 in 13 Black or African-American babies is [sic] born with sickle cell trait (SCT).” Over the past four (4) decades, the US has made significant progress in the care of people with SCD. Homer and Oyeku (2016) explain the increase in survival rate due to healthcare progress:
Even in the absence of the discovery of new medications, median survival has increased dramatically from death typically occurring during early childhood in the 1970s to survival now in the mid-50s for individuals with hemoglobin SS and mid-60s for individuals with hemoglobin SC disease. This progress has been made possible through universal newborn screening, the effective use of penicillin, and more recently of hydroxyurea, careful monitoring, and the provision of supportive care.
Testing for SCD is easy and only requires a blood test. Early testing is imperative for diagnosis and early preventative measures to prevent complications. According to the CDC (2016), the US newborn screening program requires every baby to be tested for SCD. Prior to birth, amniotic fluid can be also tested to diagnose SCD. Newborn screening is extremely important and effective so that informed parents can discuss options with their primary care doctor, a hematologist or a genetics counselor.
Find Out How UKEssays.com Can Help You!
Our academic experts are ready and waiting to assist with any writing project you may have. From simple essay plans, through to full dissertations, you can guarantee we have a service perfectly matched to your needs.
View our academic writing services
SCD disproportionately affects African-American and Hispanic communities which rely heavily on public healthcare and insurance programs. According to Hassell (2016), SCD therapies which include “coordinated care by knowledgeable providers, integrating specialized and routine health care across the life span” are not readily available to these targeted groups. A structured system of care is nonexistent for people with SCD. In addition, “mounting evidence suggests that therapies of proven benefit, including prophylactic penicillin, transcranial Doppler, and hydroxyurea therapy, are not being utilized” (Hassell, 2016).
In conclusion, one does not contract SCD, one is born with SCD. This disease is easily identified by a simple blood test and disproportionately affects those people in low-income areas. SCD treatment options are not widely available to those afflicted by the disease. The social and societal implications are far reaching and significantly negatively impact the African-American and Hispanic communities.
References
Burnes, D. P., Antle, B. J., Williams, C. C., & Cook, L. (2008). Mothers Raising Children with Sickle Cell Disease at the Intersection of Race, Gender, and Illness Stigma. Health & Social Work, 33(3), 211-220. doi:10.1093/hsw/33.3.211 Retrieved from http://go.galegroup.com.nuls.idm.oclc.org/ps/i.do?&id=GALE|A184643666&v=2.1&u=nu_main&it=r&p=AONE&sw=w&authCount=1
Centers for Disease Control and Prevention (2016). Data & Statistics. Retrieved from https://www.cdc.gov/ncbddd/sicklecell/data.html
Hassell, K. L. (2016). Sickle Cell Disease A Continued Call to Action. American Journal of Preventive Medicine. Volume 51, Issue 1, S1-S2. doi: http://dx.doi.org/10.1016/j.amepre.2015.11.002. Retrieved from http://www.ajpmonline.org/article/S0749-3797(15)00726-6/fulltext
Homer, C. J. & Oyeku, S. O. (2016). Sickle Cell Disease A Roadmap for Getting to Excellence Everywhere. American Journal of Preventive Medicine. Volume 51, Issue 1, S1-S2. doi: http://dx.doi.org/10.1016/j.amepre.2015.10.018. Retrieved from http://www.ajpmonline.org/article/S0749-3797(15)00702-3/fulltext
Howe, E. M. (2007). Untangling Sickle-cell Anemia and the Teaching of Heterozygote Protection. Science & Education, 16(1), 1-19. doi:10.1007/s11191-005-4712-7 Retrieved from http://web.a.ebscohost.com.nuls.idm.oclc.org/ehost/pdfviewer/pdfviewer?sid=a36e64b9-4230-45e0-accb-db8cdc436af1%40sessionmgr4009&vid=1&hid=4209
Liddell, C., Owusu-Brackett, N., & Wallace, D. (2014). A Mathematical Model of Sickle Cell Genome Frequency in Response to Selective Pressure from Malaria. Bulletin of Mathematical Biology, 76(9), 2292-2305. Retrieved from http://search.proquest.com.nuls.idm.oclc.org/docview/1560166043?OpenUrlRefId=info:xri/sid:primo&accountid=25320
United States Department of Health & Human Services. National Heart, Blood, and Lung Institute (2016). What is Sickle Cell Disease? Retrieved from
https://www.nhlbi.nih.gov/health/health-topics/topics/sca
Cite This Work
To export a reference to this article please select a referencing style below: