
Heterocycles1 are one of the major classes of organic compounds. They are cyclic compounds containing one or more heteroatom (oxygen, nitrogen, sulphur etc.). These compounds are of biological and industrial importance. Many materials that are essential to life include a heterocyclic moiety; examples of these are: amino acids, nucleic acids, pigments, vitamins, antibiotics and many more. Heterocycles are part of our everyday life and are present in: drugs, dyes, pesticides and plastics.2, 3
Most common heterocycles are five or six membered ring; examples of these are pyridine, pyrrole, furan and thiophene (Figure 1). It is also possible to find two or more of them fused together to give even more complex structures.4

Figure 1: Common heterocyclic structures
Many heterocyclic compounds are biosynthesised by plants and animals and hence have biological activity. These include as the haem group in the blood, and the chlorophylls ,essential for photosynthesis, both of these are essential to life.5
Examples of heterocycles used in the pharmaceutical industry include the pyridine based anti-AIDS-virus drug Nevirapine6, or the vasodilator Nicorandil for the treatment of angina (Figure 2).7
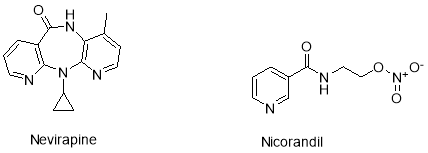
Figure 2: Example of heterocyclic drugs
Pyrimidines are an important class of heterocycles that are essential to life and biologically and pharmacologically active. Cytosine, thiamine, uracil, adenine, guanine are the nitrogen bases that are present in both DNA and RNA (Figure 3); derivatives of these have been used to form the core of drug molecules, due to their ability to selectively interact with the human body.3, 7
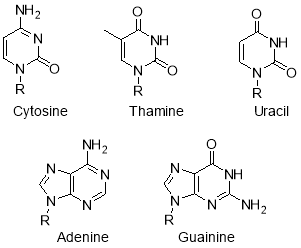
2. Synthesis of heterocycles
There are various traditional pathways leading to high yielding heterocyclic synthesis. Two of the most common are: Fischer-indole1, 8 synthesis and a Hantzsch synthesis to obtain the pyridines.
Fisher-indole synthesis (Scheme 1) has been the most important method for the preparation of substituted indoles. It involves the formation of an imine 1.3 from a hydrazine 1.1 and a ketone 1.2. This will then tautomerise to give a enamine 1.4 that will subsequently undergo a [3,3] Cope rearrangement and form a di-imine 1.5, that will then aromatise. The aromatised amine 1.6 will undergo further intramolecular cyclisation to give the indoline 1.7 which will also aromatise to give the second ring system of the indole 1.8.4, 7

Scheme 1: Fischer-indole synthesis of indoles
The Hantzsch1, 8 synthesis (Scheme 2), is a three component reaction, which firstly involves an aldol condensation of a diketone 2.1 with acetone 2.2. This is then followed by a conjugate addition of another equivalent of a diketone 2.1 to give a tetraketone 2.4. By adding ammonia it will lead to the formation of an imine and enamine component 2.5, which will subsequently react with sodium nitrate and acetic acid to aromatise to give pyridine 2.6.9

Scheme 2: Hantzsch synthesis of pyridines
These two synthetical techniques have been greatly utilised however they have their downsides; which range from the need for high temperatures, large amounts of stoichiometric reagents, as well as the poor accessibility of the starting materials. This has therefore driven research for the discovery of new methodologies for an efficient route for the synthesis of heterocycles and their derivatives.
A wide range of new heterocyclic synthetic routes using metal cross coupling reactions have arisen in particular utilising palladium as catalyst10, examples which are considered in the section below.
2.1. Synthesis of indole heterocycles using catalysis
A palladium catalysed Fisher indole synthesis has been discovered by Wagaw et. al.. It involves the cross coupling of an aryl bromide and a hydrazone. This palladium catalysed reaction has proven to be very versatile in respect to the functionality of both the reagents used. The addition of the p-toluenesulfonic acid monohydrate hydrolyses imine 3.3 therefore liberating the hydrazine which will subsequently react with a ketone to give a hydrazone. This will then undergo standard Fischer-indole process to produce 3.4 (Scheme 3).11

Scheme 3: Palladium catalysed Fischer-indole synthesis
2.1.1. Palladium catalysed reactions
Palladium catalysed reactions have been regularly used in both academic and industrial synthetic chemistry laboratories as an important method for the formation of carbon-carbon and carbon-heteroatom bonds. These have been heavily applied in the synthesis of pharmaceutically and biologically important molecules.1, 12
Many palladium catalysed reactions go through a similar catalytic cycle. The catalytic species can be formed in situ by using a palladium source such as Pd(PPh3)2Cl2 or Pd(OAc)2 with the addition of an appropriate ligand. By choosing the correct ligand this can improve two steps in the catalytic cycle; oxidative addition and reductive elimination.13
A common feature of these catalytic process (Scheme 4) is the formation of aryl/alkyl Pd(II) intermediates which will then be functionalised to form C-C or C-Heteroatom bonds that are cleaved off from the metal centre.14 Most of the Pd catalysed reactions undergo the same basic catalytic cycle involving; oxidative addition, transmetallation and reductive elimination.

Scheme 4: Palladium catalytic cycle
2.1.2. Palladium catalysed cross coupling reactions
Below in Table 1 are described some of the most common palladium catalysed reactions.15-18
Table 1: Palladium catalysed reactions
|
Name of Reaction |
Suzuki |
Stille |
Negishi |
Hiyama |
Sonogashira |
Heck |
Buckwald-Hartwig |
|
|
Catalyst |
Pd(0) |
Pd(0) |
Pd(0) |
Pd(0) |
Pd(0), Cu(I) |
Pd(0) |
Pd(0) |
|
|
Base |
Yes |
No |
No |
Yes |
Yes |
Yes |
Yes |
|
|
Reagent 1 |
|
|
|
|
|
|
|
|
|
Reagent 2 |
|
|
|
|
|
R= EWG (eg. NO2, CN, COOR) |
|
|
|
Product |
|
|
|
|
|
|
|
|














3. Recent examples of heterocyclic synthesis
There are various recent examples of interesting heterocycles synthesis. Large amounts of research covering aspects of C-H activation, atom economy improvements and efficiency with the use of different solvents has been carried out to improve the conditions of the synthesis.
An interesting class of heterocyclic moiety is pyrimidines due to their desirable biological activity. Over recent years the pyrimidine system (Figure 4) has been shown to be an important pharmacophore.19

Pyrimidines are very prevalent in nature; they are the precursors for the nucleoside bases of both DNA and RNA (Figure 3) and are also found in many more natural products such as vitamins and antibiotics. Examples, of this class of heterocycles, are shown below in Figure 5.19

Figure 5: Natural products containing pyrimidine moiety
As a result of this long-lasting interest in the pyrimidine moiety as well as in its derivatives in the use as potential drug targets, the synthesis of this heterocyclic ring has been expansively researched.15 Good yielding strategies have been developed primarily on the basis of nitrogen-carbon-nitrogen condensation reactions. An example of this is the Pinner addition (Scheme 5) of guanidines 5.2 and amidine salts to 1,3-diketones 5.1 or their derivatives.20

Scheme 5: Pinner addition for the synthesis of pyrimidines
3.1. Synthesis of β-Enaminones: an entry into pyrimidines
Gayon et. al.21 have demonstrated highly stereoselective synthesis of β-enaminones via based catalysed rearrangement of propargylic hydroxylamines. β-Enaminones are versatile compounds that have been used for their pharmacological activity, as building blocks for natural product synthesis and heterocyclic synthesis.
The initial discovery showed that propargylic hydroxylamine 6.1 underwent rearrangement to give the Cbz-protected enaminone 6.2 as a single (Z) diastereomer (Scheme 6) which could then be used for the synthesis of heterocyclic compounds such as pyrimidines.

Scheme 6: Base catalysed rearrangement reaction to afford β-enaminones
The reaction mechanism (Scheme 7) starts from the deprotonated aminol 7.2 that is easily formed; this is due to the association of the hydroxyl anions present in solution with the propargylic hydroxylamine 7.1. With the deprotonation takes place at the propargylic position an imine 7.3 is formed this is also because of the elimination of hydroxyl anions which are triggered by the deprotonation itself. Hydroxyl anions add to the imine which will eventually for the allenol 7.4. This can then undergo keto-enol tautomerisation to produce the deprotonated enaminone 7.5. The last step involves proton exchange between another molecule of propargylic hydroxylamine 7.1 and the anionic deprotonated enaminone 7.5 to give the product and another molecule of aminol 7.6.
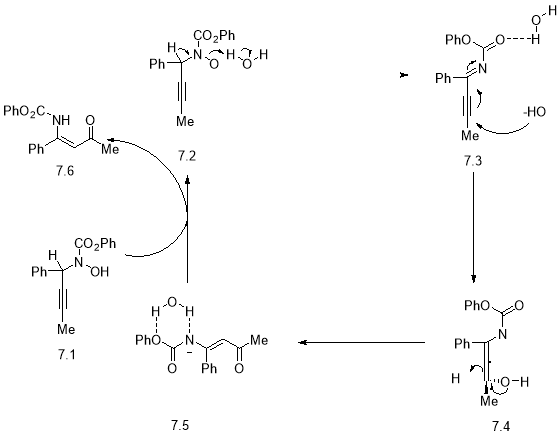
Scheme 7: Mechanism for rearrangement
These enaminones proved to be an interesting building block for the synthesis of heterocyclic compounds such as pyrimidines. The presence of a nucleophilic nitrogen atom, a double bond and an electrophilic carbonyl provided an different cyclocondensation process to simple Pinner addition on the basis of the addition of an electrophile/nucleophile partner; such as a carboxamide.
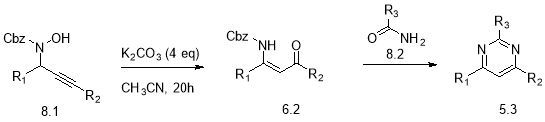
Scheme 8: Propargylic hydroxylamine to pyrimidines
The presence of the nitrogen atom on the enaminone 6.2 is advantageous for an alternative synthetic route for the synthesis of functionalised pyrimidines (Scheme 8) from readily commercially available and inexpensive carboxamides that can give easier access to pyrimidines.
3.2. Synthesis of Pyrazine Heterocycles and their Derivatives
Pyridazines have been considered one of the ‘most drugable’ heteroaromatic rings for medicinal purposes.22 Its analogues have proved to be good starting points for developing drugs for various molecular targets and have demonstrated biological activity in many key areas such as obesity, neurodegenerative diseases, inflammatory pain and many more.22
Abed et al. have recently elaborated a synthesis of novel fused pyridazines by carrying out a diaza-Wittig reaction on 1,3-diketones (Scheme 9).20

Scheme 9: Synthesis of pyrazine heterocycles
The work was divided in two parts; the first was the development of a convenient strategy to obtain versatile pyridazines containing an ester group at position 6 as a point of divergence. The latter steps involved the synthesis of pyridazines 9.4 with a ketone group at position 6. Towards this aim, different methods of cyclization techniques were applied which led to the formation of previously not known biheterocyclic compounds. This novel methodology provided an attractive synthesis for fused pyridazines derivatives (9.5, 9.6, 9.7, 9.8).20 The synthesis of functionalised pyridazines 10.4 (Scheme 10) proceeded via a diaza-Wittig reaction (10.3 to 10.4, step b.) affording a convenient and safer method for the synthesis of nitrogen heterocycles.

Scheme 10: Synthesis of pyridazines
3.3. Direct imine acylation for heterocyclic synthesis
Much attention has been focussed on the synthesis of diverse heterocyclic structure to advance the discovery of novel lead compounds for pharmaceutical discovery. A particularly useful approach is the formation of N-acyliminium ions from the acylation of imines with acid halides and anhydrides. This is a well-known reaction but very little work has previously been carried out to show the full potential of these ring closure reactions.23
Find Out How UKEssays.com Can Help You!
Our academic experts are ready and waiting to assist with any writing project you may have. From simple essay plans, through to full dissertations, you can guarantee we have a service perfectly matched to your needs.
View our academic writing services
Unsworth et. al. has provided a neat ring closing reaction (Scheme 11) by using propylphosphonic acid anhydride 11.3 (T3P) and NEt(iPr)2 for the coupling of aliphatic acids 11.2 (containing oxygen, nitrogen and sulphur nucleophiles) to imines 11.1. This will generate N-acyliminium ions 11.4 that can easily be trapped intramolecularly by the nucleophilic substituents that are present on the aliphatic acid fragment to form 11.5.

Scheme 11: Ring closing reaction for heterocyclic synthesis
This methodology has been used by Unsworth for the synthesis of the natural product evodiamine (Figure 6).24

Figure 6: Synthesis of evodiamine
4. Domino reactions
A process that involves two or more bond-forming transformations that can take place under the same reaction conditions without additional reagents and/or catalysts, are considered to be domino/tandem reaction.25
In the past decades, synthesis of heterocyclic compounds through new domino reactions has attracted many researches and is still an expanding area.26 The idea of building simple and complex heterocycles starting from very easy and reasonable building blocks using a ‘single pot’ reaction with consecutive transformation taking place, is an attractive tool for synthetic chemists, especially if the synthesis can be used to create multiple stereogenic centres.26, 27 This ‘one pot’ strategy has many advantages; reduction of solvent, waste production, reaction time and atom economy28, all of which are important for developing a more sustainable chemistry. One single reaction can potentially convert an inexpensive material to a highly complex, biologically active heterocyclic molecule.26, 29
For many years the research groups of Valotti and Arcadi have focussed their interests in developing a new synthetic route for the construction of nitrogen-containing heterocycles starting from alkyne derivatives.30 They have focused most of their attention on the synthesis of nitrogen containing heterocycles by the condensation of ketoalkynes with ammonia.30 Examples of these are shown below in Scheme 12 and show the 5-exo-dig cyclisation reactions of 4-pentynones 12.1 to synthesise polysubstituted and joined pyrrole derivatives 12.2, Ketoalkyne moiety 12.5 in an aromatic framework would allow a 6-endo-dig cyclisation of 5-acetyl-4-alkynylthiazoles 12.6 and 2-acyl-3-alkynylindoles 12.7 to pyrido[3,4-c]thiazole and pyrido[3,4-b]indoles 12.8.31, 32

Scheme 12: Synthesis of pyrrole, pyridine and indole nitrogen containing heterocycles
Another example of a tandem reaction for heterocyclic synthesis is the intermolecular 1,3-dipolar cycloaddition of nitrones for the formation of cyclic isooxazolidines (
Scheme 13). For example an interesting method has been developed for the generation of the cyclic isooxazolidines frameworks by using cheap and accessible starting material, such as Amaryllicaceae alkaloids, through a 1,2-prototropic shift of oximes.

Scheme 13: Tandem reaction for cyclic isooxazolidines
Wildman observed that the reaction of 6-hydroxybuphandidrine with hydroxylamine produced a cycloadduct; the reaction occurs by the formation of an intermediate oxime that then undergoes a subsequent 1,2-prototropic shift to give the nitrile oxide that then undergoes an intramolecular [1,3]-dipolar cycloaddition reaction.
5. Indoles
In both nature and drug discovery, a common nitrogen containing heterocycle is indole and its derivatives. These naturally occurring molecules are present in a range of compounds (Figure 7) such as amino acids (tryptophan) and hormones (melatonin) and many others.33

Figure 7: Naturally occurring indoles
As indoles are structural components of a large number of biologically active natural compounds, their synthesis and functionalization has been heavily researched, and is a crucial step in the preparation of many pharmaceutical compounds.33 Below, in Figure 8, are a couple of examples of indole containing pharmaceuticals and their applications; Sumatripan used for the treatment of migraine and Arbidol as an antiviral drug.34

Figure 8: Indole containing pharmaceutical compounds
Another potential application for indoles is the possible use of indole derived nitrones as spin traps which can be employed as free radical probes for the identification of radicals in chemical and biological systems.35, 36 The pyrroline based 5,5-dimethyl-pyrroline N-oxide (DMPO) followed by the 5-carbamoyl-5-methyl-l-pyrroline N-oxide (AMPO) have often been used as nitrene spin traps in the past years (Figure 9).37 The use of spin trapping has gained attention in the recent years and it is currently being used in the investigation of reactive intermediates in the areas of fuel cell research, nanotechnology, catalysis, environmental remediation and photodynamic therapy using electron paramagnetic resonance (EPR).38

Figure 9: DMPO and AMPO spin traps
The main disadvantages with many spin trap nitrones, for example DMPO, is the formation of secondary EPR signals; caused by the formation of other radical species caused by the instability by hydrolysis, decomposition and various other side reactions.37
Also, the slow reactivity of the superoxide radical anion to the nitrones and the short half-life of the spin adducts together, with slow rate of passive diffusion through the cell membran decreases their application in the human body as O2. – probes, unless further modifications can be carried out.38 Variants of the DMPO that contain indole moieties are 1,1,3-trimethyl-1H-isoindole-N-oxide (TMINO) and 1,1-dimethyl-3-(trifluoromethyl)-1H-isoindole-N-oxide(3-TF-TMINO) (Figure 10) which offer more stability to the oxygen radical adduct.37

Figure 10: Novel spin trap nitrones
6. Isoindolinone and Isoindoline
Novel structures related to indoles such as isoindoline and isoindolinone compounds (Figure 11) are still relatively unexplored and have only begun to be explored over the past few years.
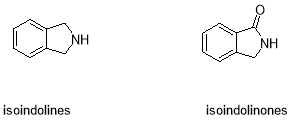
Figure 11: Isoindoline and Isoindolinone
Both of these structures have attracted a large amount of attention as pharmacophores due to their potential physiological and chemotherapeutic activity. These bicyclic models moieties have found a large importance as intermediates in the synthesis of various dugs and natural products.39
The more stable isoindolinones in particular demonstrate fascinting biological properties40, 41 as part of biologically active natural products such as magallanesine as well as drug candidates such as pagoclone shown in Figure 12.42

Figure 12: Isoindolinones as drug candidates
The biological activity of isoindolinones has been utilised for the preparation of drugs for treatments in a wide range of diseases (Figure 13) such as: diabetes treatment, anti-inflammatory, anti-hypertensive, antipsychotic agents, for the modulation of dopamine D receptor, inhibitors of amyloid protein aggregation for the treatment of Alzheimer, selective antagonist of Essential Thromobocythemia (ET) diseases related to the heart and the lungs, melanocortin subtype-4-receptor in the targeting of weight disorder and sexual dysfunction43 and as antileukemic agents.42, 44-46
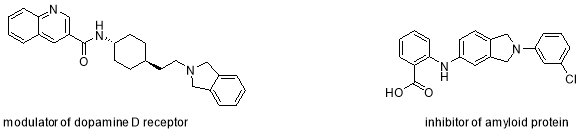

Figure 13: Isoindolines as drug candidates
There have been several methods employed traditionally for the synthesis of these compounds based on use of a wide range of transformation, such as Diels-Alder, Grignard reagents, reductions, Wittig reactions and photochemical reactions. An example of one of them follows in Scheme 14.

Scheme 14: One pot synthesis of isoindolinones
The traditional routes are usually complex and have proved to be very unsatisfactory due to the low yields and the expensive starting material.39 Most approaches do not provide a large compatibility with many functional groups and suffer from a lack of generality.38, 42 New approaches have arisen in the past decades involving palladium chemistry and lithiation procedure for the synthesis of substituted isoindolinones that have proved to be easier to handle. An example of this is shown below (Scheme 15).47

Scheme 15: Lithiation and substitution
Cite This Work
To export a reference to this article please select a referencing style below: